
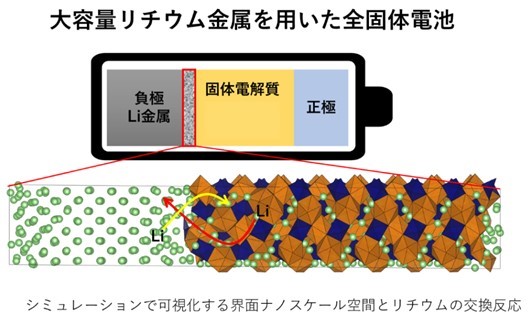
全固体リチウム電池の界面反応をシミュレーション解析 ~深層学習ポテンシャル計算による界面反応の可視化~
News&Topics
カテゴリ:プレスリリース|2024年8月21日掲載
発表のポイント
〇 深層学習ポテンシャルを用いた分子動力学シミュレーションにより、全固体電池の電極・電解質界面の原子レベルでの安定性とイオン交換反応速度を定量化することに成功。
〇 高温でも安定したLLZ固体電解質の空間電荷層の形成を確認し、リチウムイオンの移動過程を時系列で解析。
〇 様々な材料の組み合わせによる全固体電池に適用可能なシミュレーション。
〇 将来の電気自動車電源として安全性と航続距離の向上につながる全固体電池の合理的設計へ道筋。
概要
名古屋工業大学の岩崎梨音氏(研究当時:工学専攻創造工学プログラム 博士前期課程2年)、生命・応用化学類の中山将伸教授らの研究グループは、全固体電池材料(*1)で重要な電極・電解質界面について、深層学習ポテンシャルを用いた分子動力学シミュレーション(*2)を行い、原子レベルでの固体電解質の化学的安定性、界面における空間電荷層(*3)の形成、界面リチウムイオン交換反応速度(*4)の定量化などの評価に成功しました。この成果により、全固体電池の電極・電解質界面の反応機構の解明をシミュレーションで行うことが実証され、更に全固体電池の合理的設計に結び付くものと期待されます。
本研究成果は、2024年8月10日に「Communications Materials」誌に掲載されました。
研究の背景
全固体リチウム電池は、高い安全性と寿命、航続距離の延長が期待される次世代型電池として、産学で活発に研究開発が行われています。しかし、従来の液体を使った電池と異なり、主要な電極部材である正極、負極、電解質がすべて固体から構成されるため、固体と固体の接触界面におけるリチウムイオンの界面交換反応を適切に制御する必要がありました。これまでの研究によれば、界面に電気抵抗が発生しており、エネルギー変換のロスや出力特性の低下につながることが認識されています。あるいは、接触した二つの固体間での分解反応により電池が劣化する現象なども観測されてきました。しかしながら、原子レベルでどのような反応が発生しているかは長らく不明であり、シンクロトロン放射光を用いた大規模実験などによる研究などが世界的に行われています。材料シミュレーションの研究においても、スーパーコンピューターと連携した量子力学計算(*5)による、反応機構解明が取り組まれてきました。しかし、量子力学計算は現代のスーパーコンピューターをもってしても、計算負荷が大きく、固体界面におけるシミュレーションには入力できるモデルのサイズの制限や計算リソースなどに対する経済的コストなどの課題がありました。
研究の内容・成果
本研究では、従来用いてきた量子力学計算に代わり、近年普及が進んでいる深層学習ポテンシャル計算を用いて材料シミュレーションを行いました。深層学習ポテンシャルは、量子力学計算の結果データベースをあらかじめ学習することで、ユーザーが与えた結晶構造のエネルギーや原子に作用する力を即時に評価することができる手法論です。当該手法を用いて、多くの研究者が注目している金属リチウム負極とガーネット型Li7La3Zr2O12固体電解質(以下LLZ)の接触界面のシミュレーションを行いました。ソフトウェアには日本で開発されたPreferred Computational Chemistry社のMatlantis※を用いています。図1は、2022年に発表した量子力学計算(DFT計算)によりLi/LLZ界面の研究を行ったモデルであり、当時は298原子による評価を行いました(R. Iwasaki et al. Phys. Stat. Sol. B, 259, 2100546 (2022) DOI: 10.1002/pssb.202100546)。本研究では、1024原子のモデルに拡張し、更にDFT計算では困難であったリチウムイオンの移動過程についても検討しました。
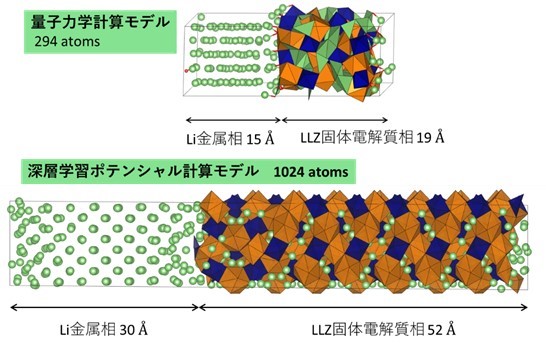
図1 従来用いられる量子力学計算に使用したモデルと、本研究で用いた深層学習ポテンシャル計算に使用したモデル。深層学習ポテンシャルを用いると計算が高速に行えるため、固体電解質を十分厚くした状況で評価が可能。
シミュレーションの結果、ガーネット型LLZ固体電解質は高温(1000℃)でも金属リチウムと分解反応する挙動は観測されず、非常に安定な固体電解質材料であることが確認されました。また、リチウムイオンの移動過程を時系列解析した結果、固体電解質の表面に正電荷を有するリチウムイオンが組成式(Li7La3Zr2O12)よりも過剰に堆積した状態が形成することが分かりました。LLZ材料にて正電荷が過剰に堆積した空間の形成した結果、空間電荷層が形成したものと考えられます。実際に図3に示すように、深層学習ポテンシャルによる1ナノ秒の分子動力学計算の結果を用いて、電子構造を可視化したものを図2に示します。この図からは、Li/LLZ界面近傍で、LLZ相のバンドが1.1 nm にわたって折れ曲がっている結果が得られ、空間電荷層の存在が確認されました。空間電荷層の形成は、Li金属からLLZ相に電子が移動し還元分解を抑制するため、固体電解質の安定性を向上させることにつながります。さらに、界面におけるリチウムイオン交換反応速度も、シミュレーションで定量化することができました。LLZ相からLi相、Li相からLLZ相へのリチウムイオンの移動の活性化障壁エネルギーは160meVおよび88meVであり非常に低い値を示しました。界面でのリチウムイオンの交換はLi/LLZ界面では非常にスムーズであり、固体内のイオンの拡散が律速していることが定量的に確認されました。
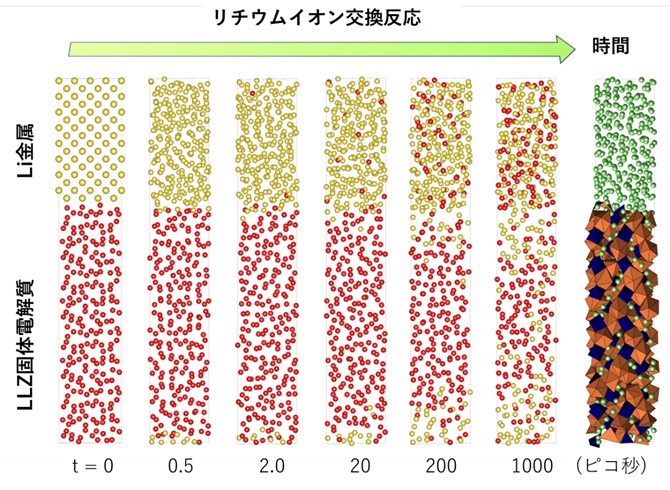
図2 LLZ固体電解質とLi金属の間で移動するLiを時系列に可視化した図。時刻0の時に、Li金属相にあるLiを黄色、LLZ固体電解質相にあるLiを赤色で表しており、時間が経過するにつれて、2つの相のリチウムイオンが混ざり合っていくことが分かる。
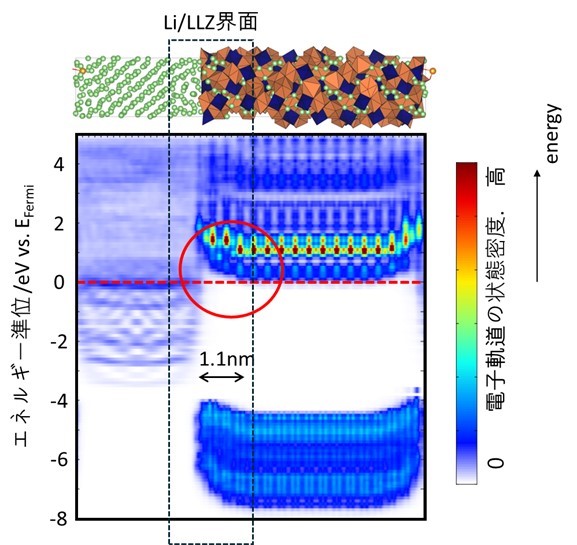
図3 深層学習ポテンシャルで得られたLi/LLZ界面モデル構造を用いて、量子力学計算により再計算を1回行い、電子状態解析を行った結果。Li/LLZ界面近傍(黒破線枠)において、赤丸で囲んだLLZ相の伝導帯バンドが屈曲しており、空間電荷層が形成されたことが示された。
社会的な意義
本研究成果によって、蓄電池材料の界面の安定性、リチウムイオン交換反応速度などが定量的にシミュレーションできることが分かりました。この成果はLi/LLZ界面にとどまらず多くの電池材料に当てはめることができます。また、実験で得られる複雑なスペクトルの解釈に悩むことなく、直観的に界面で発生している反応を可視化することもでき、全固体電池設計のための研究を加速させることが期待されます。
今後の展望
本研究成果は、負極と固体電解質のみではなく、正極と固体電解質の反応性評価にも用いることができます。また、電極と固体電解質間ではしばしば副反応が発生し、コート材を導入するなどのプロセスが必要なことが経験的に知られていますが、原子レベルでコート材がどのような役割を果たすかはいまだに不明な点が多い状況です。全固体電池で経験的に用いられている技術やノウハウについても、体系的解明に近づくことが期待されます。体系的な界面設計指針が得られることで、膨大な組み合わせが存在する電極・電解質・コート材などの選定をシミュレーションやインフォマティクスで行うことが可能となり、電気自動車電源やスマートグリッド用電源となる安全かつ大容量全固体電池の実現に結びつきます。
本研究は、科研費新学術領域研究・蓄電固体界面(19H05815)および文部科学省・DX-GEMプログラム(.JPMXP1122712807)等の支援を受けて実施されました。
用語解説
(*1)全固体電池材料
従来のリチウムイオン電池はLi塩を溶かした有機電解液を用いており、火災・爆発などのリスクを抱えています。全固体電池は、電解液をセラミックスでできた固体電解質に置き換えた電池で、安全性に優れています。さらに、これまでを使用できなかった金属Liを負極に用いることが可能であり、電池容量の大容量化にもつながると期待されています。
(*2)深層学習ポテンシャルを用いた分子動力学シミュレーション
深層学習ポテンシャルは、量子力学計算のデータベースを学習させ、粒子間に働く力やエネルギーを短時間で計算するAI技術です。また、ニュートン方程式に基づいて、原子の位置や速度の時間変化をシミュレーションする手法を分子動力学法といいます。深層学習ポテンシャルを用いることで、粒子に働く力を高速に評価し、効率的に分子動力学法のシミュレーションが実施できるようになりました。
(*3)空間電荷層
一定空間に、電荷が溜まった領域のこと。本研究の場合、Li金属とLLZ固体電解質材料の界面で、局所的にリチウムイオン(正電荷)が負電荷に比べて過剰にあり、空間電荷層が形成されています。
(*4)界面リチウムイオン交換反応
全固体電池を構成する正極と固体電解質、または負極と固体電解質の間で、常にリチウムイオンが2つの材料の界面を通過して行き来している反応を界面リチウムイオン交換反応と言います。交換反応の速度が速いと、界面での電気抵抗が減少し、高い出力でも電池が動作するようになります。
(*5)量子力学計算
量子力学方程式により電子の波動関数を求め、原子に作用する力や入力した分子や結晶のエネルギー、電子構造などを評価することができる計算法を量子力学計算と言います。
※各社のサービス等の登録商標です。
論文情報
論文名:Universal-neural-network-potential molecular dynamics for lithium metal and garnet-type solid electrolyte interface
著者名: R. Iwasaki, N. Tanibata, H. Takeda, M. Nakayama*(*責任著者)
掲載雑誌名:Communications Materials
公表日:2024年8月10日
DOI:10.1038/s43246-024-00595-0
URL:https://doi.org/10.1038/s43246-024-00595-0
お問い合わせ先
研究に関すること
名古屋工業大学 生命・応用化学類
教授 中山 将伸
TEL: 052-735-7273
E-mail: nakayama.masanobu[at]nitech.ac.jp
広報に関すること
名古屋工業大学 企画広報課
TEL: 052-735-5647
E-mail: pr[at]adm.nitech.ac.jp
*[at]を@に置換してください。
実用的な高エネルギー密度のコバルト・ニッケルフリー電池材料を開発―ナノ構造を高度に制御したリチウムマンガン酸化物材料の合成に成功― 文部科学省「情報ひろば」企画展示室において簡易住宅(インスタントハウス)の企画展示を開催